Type 2 diabetes is one of obesity’s most tightly linked comorbidities, and its prevalence is rising in parallel with the global obesity epidemic. Uncontrolled obesity often leads to insulin resistance, beta cell failure, and hyperglycemia. Although effective, many glucose-lowering agents circumvent rather than address the pathophysiology of this disease. The thiazolidinediones (TZDs) are a class of anti-diabetic drugs that are unique as they are insulin sensitizers capable of delaying or even preventing the onset of diabetes. Despite potent action on insulin sensitization, clinical use of TZDs has declined due to a number of serious adverse side effects. As ligands for the nuclear receptor peroxisome proliferator activated receptor-γ (PPARγ), the therapeutic actions of TZDs have been assumed to result from their classical agonism towards PPARγ to drive adipocyte differentiation. That is, TZDs are thought to act through the formation of new, healthy adipocytes can effectively store excess energy in the adipose tissue. PPARγ is highly expressed in adipose tissue, where it was first characterized as a master regulator of adipocyte differentiation and function. Activation of PPARγ improves insulin sensitivity owing to diverse roles on both adipocyte and non-adipocyte cells, including macrophages, T-cells, muscle, and others. PPARγ is intimately linked to insulin sensitivity, and genetic variants in the PPARG gene locus are found to associate with diabetes risk.
Our work:
Post-translational modification of PPARγ
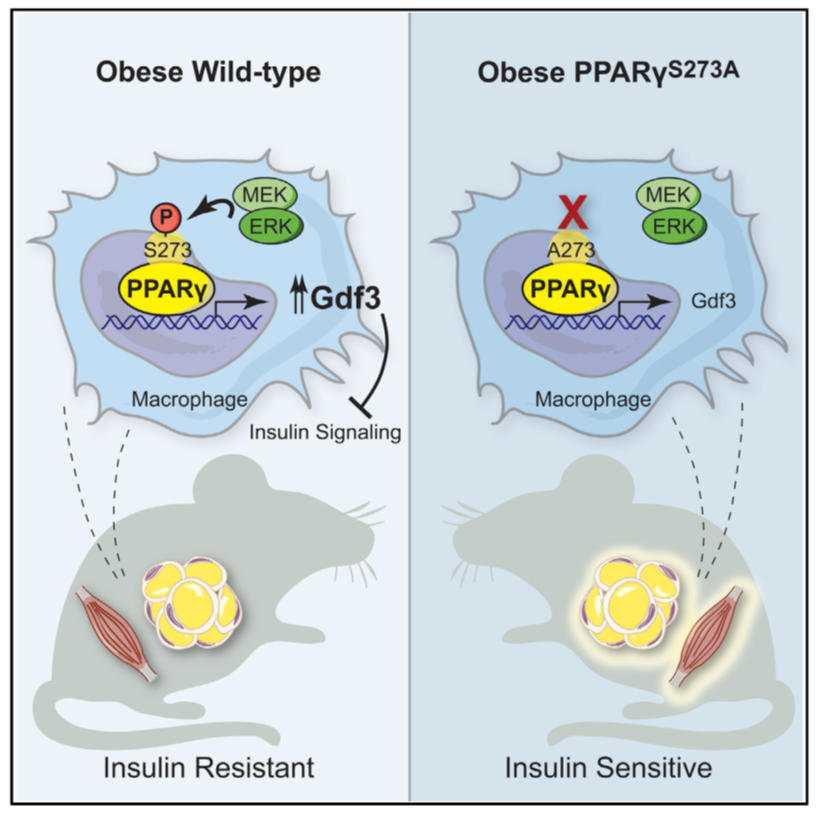
Phosphorylation of PPARγ at serine 273 (p-S273) is observed in adipose tissue shortly after the initiation of high-fat diet feeding and levels continue to increase with progressive obesity. When diabetic patients or mice are treated with TZDs, the phosphorylation of PPARγ S273 is blocked, gene expression profiles are normalized, and insulin sensitivity improves (Nature 2010). Furthermore high-affinity ligands of PPARγ designed to only block p-S273, still retain anti-diabetic effects but lack activity on adipogenesis (Nature 2011). These “non-agonist ligands” are safe and do not trigger the side effects associated with TZDs use. Likewise, in vivo inhibition of the kinase responsible for PPARγ S273 phosphorylation, extracellular signal-regulated kinase (ERK), improves insulin sensitivity in obese animals (Nature 2015, Mol Metabolism 2018) . These experiments had suggested a causal role for PPARγ p-S273 to promote insulin resistance. We firmly establish this causal link with a strain of mice (PPARγ Serine 273 to Alanine global knock-in, PPARγS273A/S273A) which cannot be phosphorylated at S273 and also retain insulin sensitivity (Cell Metabolism 2020). Our model demonstrates that PPARγ p-S273 is sufficient to impart obesity-mediated insulin resistance. Quite unexpectedly, we observe a highly specific downregulation of Growth Differentiation Factor 3 (Gdf3) in epididymal white adipose tissue from PPARγS273A/S273A mice. These findings suggest that one of the actions of TZDs is to targeting PPARγ S273 phosphorylation, downregulate GDF3 levels, and strongly improve insulin sensitivity.
Gdf3: the missing piece. GDF3 is a member of the TGFb superfamily and a ligand for the ALK7 type-1 activin receptor. ALK7 signaling affects adipose tissue lipolysis through transcriptional regulation of beta-adrenergic receptor expression. Considerable controversy exists surrounding what role GDF3 plays in differentiated cells. GDF3 is highly expressed in stem cell populations and helps maintain pluripotency. However, Gdf3 mRNA levels decline sharply after differentiation. Due to the critical role of GDF3 in embryonic development, mice with germline deletion of Gdf3 exhibit decreased survival and developmental abnormalities. While these models are protected from diet-induced obesity, the mechanistic basis for these phenotypes is challenging to interpret.
Gdf3 mRNA levels significantly increase in obesity, aging, inflammation, and following an ischemic event. To understand the role of GDF3 in regulating these diseases, we need to understand its biological functions and mechanisms. We developed gain- and loss-of function models in adult mice to avoid confounding concerns of the role of GDF3 in development.

|
|
Levels of Gdf3 mRNA are controlled by phosphorylation of PPARg S273. Phosphorylation of PPARγ S273 is increased in obesity and correlates with insulin resistance in patients and in mice. Gdf3 mRNA levels also correlate with PPARγ S273. A) Decrease Gdf3 mRNA is seen in epididymal white adipose tissue (eWAT) from obese mice with an insulin-sensitizing homozygous genetic knock in of a non-phosphorylatable allele (PPARγ S273). B) Decreased Gdf3 mRNA is also observed in obese wildtype mice following treatment with Rosiglitazone (Rosi), a PPARγ ligand that sterically blocks S273 phosphorylation or pharmacological inhibitors of the PPARγ kinases (MEK kinase inhibitor, MEKi 1, PD0325901; or MEKi2, Trametinib). Whole-body insulin sensitivity is observed in all models where GDF3 levels are reduced (Hall et al., 2020). C) Gdf3 mRNA increases in eWAT of mice on HFD and in D) subcutaneous adipose tissue of patients with obesity. |
Exogenously increasing GDF3 expression with adeno-associated virus (AAV) is sufficient to make even lean mice moderately insulin resistant. Our loss of function model uses inducible whole-body Gdf3 knockouts to examine the effects of GDF3 loss of function after the onset of diet-induced obesity. Our findings show that mice with lower levels of GDF3 show improved glucose tolerance due to reduced levels of lipolysis. Loss of GDF3 as an ALK7 receptor agonist should produce lower fat mass and insulin resistance. Instead, we find that GDF3 loss of function has the opposite effect—lack of GDF3 in obese mice decreases lipolysis and promotes insulin sensitivity without affecting body weight. Conversely, GDF3 gain of function increases rates of lipolysis and insulin resistance. We find that while GDF3 may signal through the ALK7 (ACVR1c) type 1 receptor, it also strongly activates another type 1 receptor ALK5 (TGFBR1). Detailed investigation of GDF3 signaling revealed that GDF3 can simultaneously inhibit BMP-derived SMAD1/5/8 signaling and stimulate activin-like SMAD2/3 signaling in vitro, ex-vivo, and in vivo (Nature Communications 2025). Overall, our study provides new insights into the role of GDF3 in maintaining metabolic homeostasis. We are continuing to explore the mechanistic basis for these phenotypes.